
Từ cuối tháng 5 năm 2021, các nhà khoa học Mĩ - Úc đã đưa ra phát hiện như vậy. Có nghĩa là, vùng Đông Á đã tiếp xúc sâu sắc với ông tổ của Covid-19 từ hơn 2 vạn năm trước rồi !
Ông tổ cách này hơn 2 van năm của người Việt chúng ta (và người Nhật Bản, người Trung Quốc) đã chung sống với ông tổ của Covid-19 rồi !
Đấy là ý chính của phát kiến khoa học lần này. Nó làm chúng ta nhớ đến phát kiến của một số năm về trước, rằng: tổ tiên của loài người thực sự là một con đười ươi và một con lợn, gen của hai con ấy trộn vào nhau và bây giờ còn lưu giữ trong mỗi chúng ta. Xem lại phát kiến đó trên Giao Blog ở đây (năm 2013).
Dưới là tin tức về bộ "siêu gen" của người Đông Á chúng ta. Đông Á cũng là vùng văn hóa chữ Hán.
Đi dần tư liệu lên.
Tháng 7 năm 2021,
Giao Blog
---
東アジア、2万年以上前にコロナ流行の痕跡 ゲノム研究で発見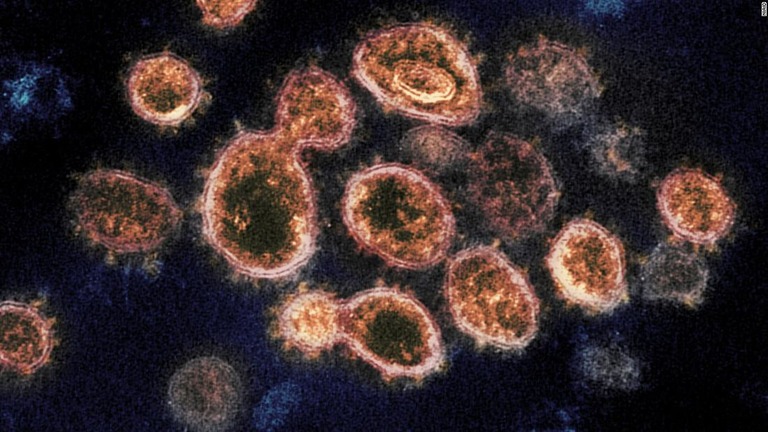
(CNN) 東アジアで2万年以上前にコロナウイルスが流行していたとする研究結果が報告された。米豪共同の研究チームが世界各地に住む人々の全遺伝情報(ゲノム)を解析し、米科学誌カレント・バイオロジーに論文を発表した。
24日に発表された論文によると、世界26カ所で計2500人あまりのゲノムを解析した結果、東アジアに住む人々の遺伝子に、コロナウイルスとの最も古い接触を示す痕跡が見つかった。
チームの研究者によると、ウイルスは一般に人間の細胞に乗っ取って増殖し、人間側の遺伝子はこれに応じて変化を起こす。最近の研究で、その痕跡を数万年前までさかのぼって検出できるようになった。
コロナウイルスに関連する変化は中国と日本、ベトナム国内の計5カ所で見つかった。チームは、各地で別々に起きた流行が東アジア全体に広がったとの見方を示す。
インフルエンザのように季節性のある流行だったのか、新型コロナウイルスのように通年発生していたのかは明らかでないという。
感染を経験して遺伝子が変化した集団は、コロナウイルスに打ち勝つ力をつけて生存競争に有利となり、長い年月を経て人口全体に占める割合が大きくなったと考えられる。
https://www.cnn.co.jp/fringe/35172994.html
..
Article

Keywords
Introduction
Coronaviruses have been behind three major zoonotic outbreaks.1 The first outbreak, known as SARS-CoV (severe acute respiratory syndrome coronavirus), originated in China in 2002 and infected more than 8,000 and killed more than 800 people.2 Four years later, MERS-CoV (Middle East respiratory syndrome coronavirus) affected >2,400 and killed over 850 people (https://www.who.int). The most recent outbreak began in late 2019 when SARS-CoV-2 emerged in China, triggering an ongoing pandemic (coronavirus disease 2019 [COVID-19]).3
The research on SARS-CoV-2 epidemiology has revealed that socioeconomic (e.g., access to healthcare, testing, and exposure at work), demographic, and personal health factors all play a major role in SARS-CoV-2 epidemiology.4, 5, 6 Additionally, several genetic loci that mediate SARS-CoV-2 susceptibility and severity have been found in contemporary European populations,7, 8, 9, 10 one of which contains a genetic variant that increases SARS-CoV-2 susceptibility that likely increased in frequency in the ancestors of modern Europeans after interbreeding with Neanderthals.11
Throughout the evolutionary history of our species, positive natural selection has frequently targeted proteins that physically interact with viruses—e.g., those involved in immunity or used by viruses to hijack the host cellular machinery.12, 13, 14 In the millions of years of human evolution, selection has led to the fixation of gene variants encoding virus-interacting proteins (VIPs) (Data S1A) at three times the rate observed for other classes of genes.13,15 Strong selection on VIPs has continued in human populations during the past 50,000 years, as evidenced by VIP genes being enriched for adaptive introgressed Neanderthal variants and also selective sweep signals (i.e., selection that drives a beneficial variant to substantial frequencies in a population), particularly around VIPs that interact with RNA viruses (Data S1B), a viral class that includes the coronaviruses.16,17
The accumulated evidence suggests that ancient RNA virus epidemics have occurred frequently during human evolution; however, we currently do not know whether selection has made a substantial contribution to the evolution of human genes that interact more specifically with coronaviruses.
Accordingly, here, we investigate whether ancient coronavirus epidemics have driven past adaptation in modern human populations, by examining whether selection signals are enriched within a set of 420 VIPs that interact with coronaviruses (denoted CoV-VIPs; Data S1C) across 26 human populations from the 1000 Genomes Project.18 These CoV-VIPs comprise 332 SARS-CoV-2 VIPs identified by high-throughput mass spectrometry (Data S1D),19 and an additional 88 proteins that were manually curated from coronaviruses literature (e.g., SARS-CoV-1, MERS, HCoV-NL63, etc.; Data S1C)16 and are part of a larger set of 5,291 VIPs (STAR Methods; Data S1A) from multiple viruses.16 Our focus on VIPs is motivated by evidence indicating that these protein interactions are the central mechanism that viruses use to hijack the host cellular machinery.16,19 Accordingly, VIPs are much more likely to have functional impacts on viruses than other proteins (STAR Methods). An alternative that we cannot exclude however is that a different type of virus that happens to use similar VIPs as coronaviruses might have driven adaptation signals at CoV-VIPs.
Our analyses find a strong enrichment in sweep signals at CoV-VIPs across multiple East Asian populations, which is absent from other populations. This suggests that an ancient coronavirus epidemic (or another virus using similar VIPs) drove an adaptive response in the ancestors of East Asians. Further, by leveraging ancestral recombination graph approaches,20,21 we find that 42 CoV-VIPs may have come under selection around 900 generations (∼25,000 years) ago and exhibit a coordinated adaptive response. We further show that the CoV-VIP genes are enriched for anti- and proviral effects and variants that affect COVID-19 etiology in the modern British population (https://grasp.nhlbi.nih.gov/Covid19GWASResults.aspx).22,23 We further show that the inferred underlying causal mutations are situated near to regulatory variants active in lungs and other tissues impacted by COVID-19. These independent lines of evidence support an ancient coronavirus (or a similarly interacting virus) epidemic that emerged in the ancestors of contemporary East Asian populations.
Results
Signatures of adaptation to an ancient epidemic
Viruses have exerted strong selective pressures on modern humans.15,17 Accordingly, we use two statistical tests that are sensitive to such genetic signatures (i.e., selective sweeps)—nSL24 and iHS25—while being insensitive to background selection.26,27
After scanning each of the 26 populations for selection signals, we apply an enrichment test that was previously used to detect enriched selection signals in RNA VIPs in human populations.17 Briefly, for each population and selection statistic, we rank all genes based on the average selection statistic score observed in genomic windows ranging from 50 kb to 2 Mb (STAR Methods). Different window sizes are used because smaller windows tend to be more sensitive to weaker sweeps, whereas larger windows tend to be more sensitive to stronger sweeps (STAR Methods).17 After ranking the gene scores, we estimate an enrichment curve (Figure 1) for gene sets ranging from the top 10 to 10,000 ranked loci (STAR Methods). The significance of the whole enrichment curve is then calculated using a genome block-randomization approach that accounts for the genomic clustering of neighboring CoV-VIPs and provides an unbiased false-positive risk (FPR) for the whole enrichment curve28 by re-running the entire enrichment analysis pipeline on block-randomized genomes (STAR Methods).17 For our control gene set, we use protein-coding genes situated at least 500 kb from CoV-VIPs to avoid overlapping the same sweep signals. Additionally, genes in the control sets are chosen to have similar characteristics as the CoV-VIPs (e.g., similar recombination, density of coding sequences, etc.; see STAR Methods for the complete list of factors) to ensure that any detected enrichment is virus specific rather than due to a confounding factor.17 Finally, we also exclude the possibility that functions other than viral interactions might explain our results by running a Gene Ontology analysis (STAR Methods; Data S1E and S1F; Figures S1A and S1B).29

Figure 1. Coronavirus VIPs nSL ranks enrichment
(A)–(E) are East Asian populations, and (F)–(I) are populations from other continents. The y axis represents the bootstrap test (STAR Methods) relative fold enrichment of the number of genes in putative sweeps at CoV-VIPs, divided by the number of genes in putative sweeps at control genes matched for multiple confounding factors. The x axis represents the top rank threshold to designate putative sweeps. Black full line, average fold enrichment over 5,000 bootstrap test control sets. Fold enrichments greater than 20 are represented at 20. Gray area, 95% confidence interval of the fold enrichment over 5,000 bootstrap test control sets. The rank thresholds where the confidence interval lower or higher fold enrichment has a denominator of zero are not represented (for example, graph B, top 10 rank threshold). Lower confidence interval fold enrichments higher than 20 are represented at 20 (for example, graph B, top 30 rank threshold). Red dots, bootstrap test fold enrichment p < 0.001. Orange dots, bootstrap test fold enrichment p < 0.05. Note that the bootstrap test p values are not the same as the whole curve enrichment false positive risk (FPR) estimated using block-randomized genomes on top of the bootstrap test (STAR Methods). Related to STAR Methods and Figures S2–S4.
Applying this approach to each of the 26 populations from the 1000 Genomes Project dataset, we find a strong enrichment of sweep signals in CoV-VIPs that is specific to the five East Asian populations (whole enrichment curve for nSL and iHS combined FPR = 2.10−4; Figures 1 and S2A–S2N; STAR Methods). No enrichment is observed for populations from other continents, including in neighboring South Asia (whole enrichment curve for nSL and iHS combined FPR > 0.05 in all cases; Figures 1 and S2F–S2I). Further, no enrichment is detected for VIP sets for 17 other viruses in East Asian populations (whole enrichment curve for nSL and iHS separately or combined; p > 0.05 in all cases; Figures S3 and S4). Taken together, these results suggest that coronaviruses (or a virus interacting similarly with hosts) have driven ancient epidemics in East Asia. This enrichment is unlikely to have been caused by any other virus represented in our set of 5,291 VIPs (Data S1A), but we still cannot exclude that a currently unknown type of virus that happened to use similar VIPs as coronaviruses could have been involved instead. The enrichment is most substantial for the top-ranked gene sets ranging between the top 10 and top 1,000 loci (Figure 1; whole enrichment curve FPR = 3.10−6 for nSL, FPR = 4.10−3 for iHS, and FPR = 6.10−5 for iHS and nSL combined) and is particularly strong for the top 200 loci in large windows (1 Mb) where a 4-fold enrichment is observed for both nSL and iHS statistics (pertaining to between 10 and 13 selected CoV-VIPs among the top 200 ranked genes; Data S1G). This suggests strong selection at multiple CoV-VIPs. That the selected haplotype structures are detected by both the iHS and nSL statistics suggests that they are unlikely to have occurred prior to 30,000 years ago, as both statistics have little power before this time point.30
An ancient epidemic in the ancestors of East Asians starting more than 20,000 years ago
To further test the existence of an ancient viral epidemic in East Asia, we use a recent ancestral recombination graph (ARG)-based method, Relate,20 to infer the timing and trajectories of selected loci for the CoV-VIPs. If the selective pressure responsible for the multiple independent selection events at CoV-VIPs was sudden, as expected from a new epidemic, these selection events should have started independently around the same time. By estimating ARGs at variants distributed across the entire genome, Relate can reconstruct coalescent events across time and detect genomic regions impacted by positive selection. To approximate the start time of selection, Relate estimates the first historical time point that a putatively selected variant had an observable frequency unlikely to be equal to zero (STAR Methods). We use this approximation as the likely starting time of selection (STAR Methods). Additionally, we use the iSAFE software31—which enables the localization of selected variants—along with a curated set of regulatory variants (expression quantitative trait loci [eQTLs]) from the GTEx Project32 to help identify the likely causal mutations in the selected CoV-VIP genes. There is good evidence that most adaptive mutations in the human genome are regulatory mutations.26,33, 34, 35 Accordingly, we find that iSAFE peaks are significantly closer to GTEx v8 eQTLs proximal to CoV-VIP genes than expected by chance (iSAFE proximity test; p < 10−9; STAR Methods). Therefore, for each CoV-VIP gene, we choose a variant with the lowest Relate p value (<10−3; STAR Methods) that is situated at or close to a GTEx eQTL associated with the focal gene to estimate the likely starting time of selection for that gene (STAR Methods; Figure S5A).
Using this approach, we observe 42 CoV-VIPs (Data S1H; Figure S5A) with selection starting times clustered around 870 generations ago (∼200 generations wide, potentially due to noise in our estimates; Figure 2). While this amounts to about four times more selected CoV-VIP genes than were detected using either nSL or iHS (both detected around ten CoV-VIPs among the top 200 ranked genes; Data S1G), this is not unexpected, as Relate has more power to detect selection events than nSL and iHS when the beneficial allele is at intermediate frequencies (typically <60%; Figure 3; see Enard and Petrov,17 Ferrer-Admetlla et al.,24 and Voight et al.25). The tight clustering of starting times forms a highly significant peak (peak significance test p = 2.3.10−4; Figure 2) when comparing the observed clustering of CoV-VIPs start times with the distribution of inferred start times for randomly sampled sets of genes (STAR Methods). Further, this significance test is not biased by the fact that CoV-VIPs are enriched for sweeps, as the test remains highly significant (p = 1.10−4) when using random control sets with comparable high-scoring nSL statistics (STAR Methods). Thus, the tight temporal clustering of selection events is a specific feature of the CoV-VIPs, rather than a confounding aspect of any gene set similarly enriched for sweeps.

Figure 2. Timing of selection at CoV-VIPs
The figure shows the distribution of selection start times at CoV-VIPs (pink distribution) compared to the distribution of selection start times at all loci in the genome (blue distribution). Details on how the two distributions are compared by the peak significance test, and how the selection start times are estimated with Relate, are provided in STAR Methods. Related to STAR Methods and Figure S1.

Figure 3. Selected CoV-VIPs allele frequency trajectories over time estimated by CLUES in East Asia
Each frequency trajectory is for one of the 42 Relate selected mutations at CoV-VIPs within the peak around 900 generations ago (STAR Methods).
(A) Frequency trajectories in the Chinese Dai CDX 1000 Genomes population.
(B) Same but zoomed in from frequencies 0%–10%.
(C) Frequency trajectories in the Han Chinese from Beijing CHB 1000 Genomes population.
(D) Same but zoomed in from frequencies 0%–10%.
Related to STAR Methods.
Consequently, our results are consistent with the emergence of a viral epidemic ∼900 generations, or ∼25,000 years (28 years per generation),36 ago that drove a burst of strong positive selection in East Asia. Selection events starting 900 generations ago clearly predate the estimated split of different East Asian populations included in the 1000 Genomes Project from their shared ancestral population.18
Although selective pressures other than a coronavirus or another unknown type of virus with similar host interactions might also contribute to these patterns, we note that the signal is restricted specifically at CoV-VIPs and none of 17 other viruses that we tested exhibit the same temporal clustering (peak significance test p > 0.05 in all cases; STAR Methods). Further, this test remained highly significant when retesting the clustering of CoV-VIPs using only RNA VIPs as the control set (p = 4.10−4; Data S1B). Importantly, the estimate of an ancient viral epidemic starting ∼25,000 years ago in East Asia is remarkably congruent with the 23,000 years estimate for the emergence of sarbecoviruses (the viral family of SARS-CoV-2).37
Strong selection drove coordinated changes in multiple CoV-VIP genes over 20,000 years
To learn more about the start and duration of selection acting in East Asia, we use CLUES21 to infer allele frequency trajectories and selection coefficients for the inferred beneficial mutations proximal to the 42 CoV-VIP genes with selection starting 900 generations ago according to Relate (Figure 3). We anticipate that selection was probably strongest when the naive host population was first infected, before gradually waning as the host population adapted to the viral pressure.38 Similarly, a decrease in the virulence of the virus over time, a phenomenon that has been reported during long-term bouts of host-virus coevolution,39 would also result in the gradual decrement of selection over time. Hence, for each of the 42 CoV-VIPs predicted to have come under selection ∼900 generations ago, we use CLUES to estimate the selection coefficient in two successive time intervals (between 1,000 and 500 generations ago and from 500 generations ago to the present), predicting that selection would be stronger in the oldest interval. We note that a 500 generations interval was reported as the approximate time span that CLUES provides reliable estimates for humans.21 Following the protocol of Stern et al.,40 we base our estimates on two of the five East Asian populations (i.e., Dai and Beijing Han Chinese; Figures 3A and 3B and 3C and 3D, respectively).
CLUES infers more complex frequency trajectories than an abrupt jump in frequency 900 generations ago. Instead, the estimated trajectories (Figures 3A–3D) suggest that 900 generations ago is the approximate time when the bulk of the selected variants reached a frequency of a few percent or more and when there is an acceleration in the frequency increase (Figures 3B and 3D). Note that this does not contradict the strong peak of selection times starting around 900 generations ago found by Relate, as this is the time when Relate estimates frequencies clearly distinguishable from zero (STAR Methods). This might correspond to the transition between the establishment and exponential phases of the sweeps and might imply that the selective pressure could be older than 900 generations. Although the slow starts of frequency increases make it hard to pinpoint when selection started exactly, the vast majority of the selected alleles appear to have reached 5% or higher frequencies by 600 generations, thus making it highly unlikely that the selection would have started later. Frequency trajectories estimated in the Yoruba African population (Figure 4A) or the British European population (Figure 4B) also show very low initial frequencies. The selected variants in East Asia are found nowadays at very low frequencies, especially in Africa (Data S1I).

Figure 4. Selected CoV-VIPs allele frequency trajectories over time estimated by CLUES in Africa (Yoruba) and Europe (British)
Same as Figure 3.
(A) Yoruba population. The graph includes 17 frequency trajectories, the 25 other alleles selected in East Asia being absent in the Yoruba sample (but not Africa overall; see Data S1I).
(B) British population. The graph includes 35 frequency trajectories, the other seven alleles selected in East Asia being absent in the British sample.
Related to STAR Methods.
The selected mutations are estimated to have continually increased in frequency in East Asia until ∼200 generations (∼5,000 years) ago (Figures 3A and 3C). Accordingly, CLUES estimates high selection coefficients between 1,000 and 500 generations ago (Dai average s = 0.034; Beijing Han average s = 0.042; Figures 5A and 5B) but much weaker selection coefficients from 500 generations ago to the present (Dai average s = 0.002; Beijing Han average s = 0.003; Figures 5A and 5B). These patterns are consistent with the appearance of a strong selective pressure that triggered a coordinated adaptive response across multiple independent loci, which waned through time as the host population adapted to the viral pressure and/or as the virus became less virulent.

Figure 5. Coronavirus selected VIPs selection coefficients estimated by CLUES
This figure shows classic R boxplots of selected coefficients at the 42 Relate selected mutations within the peak around 900 generations ago (STAR Methods).
(A) Selection coefficients in the Chinese Dai CDX 1000 Genomes population.
(B) Selection coefficients in the Han Chinese from Beijing CHB 1000 Genomes population. Left: average selection coefficients between 0 and 500 generations ago are shown. Right: average selection coefficients between 500 and 1,000 generations ago are shown.
Related to STAR Methods.
Validation of direct physical interactions between selected COV-VIPs and SARS-CoV-2 proteins
To further validate that an ancient viral epidemic was responsible for the observed selection signals, next we test whether the 35 out of 42 selected CoV-VIPs that interact with SARS-CoV-2 (as opposed to other coronaviruses in our dataset) are indeed CoV-VIPs and directly interact with SARS CoV-2 viral proteins. While these interactions were originally identified by high-throughput mass spectrometry,19 high-throughput mass spectrometry can sometimes identify indirect interactions in a larger protein complex or false positives altogether.41 We co-express the candidate CoV-VIPs:SARS CoV-2 protein pairs in a cell-free protein expression system and test their interactions using an AlphaLISA protein:protein interaction assay (STAR Methods). This approach (Figure S6A) was previously used for rapid analysis of intra-viral PPI network of Zika virus.42 The assay is expected to detect ∼70% of protein interactions with human proteins (30% false negative rate; STAR Methods). Out of 35 selected SARS-CoV-2 CoV-VIPs, 33 interacting protein pairs can be tested with the assay (STAR Methods). Figure 6 highlights the results for six of the 33 CoV-VIPs, while Figure S6 presents the results for the remaining CoV-VIPs. Among the 33 interactions tested, we confirm 24 or 73%, the expected confirmation rate (taking the false negative rate into account) if 100% or close to 100% of the selected CoV-VIPs are indeed CoV-VIPs (Figures 6A–6C and S6B; Data S1J). This very high validation rate further strengthens the evidence for an ancient viral epidemic in East Asia.

Figure 6. Validation of selected CoV-VIPs/SARS-CoV-2 protein interactions using cell-free expressed proteins
(A) A representative image of SDS-PAGE gel loaded with in vitro translation reactions co-expressing human VIPs/SARS-CoV-2 proteins in Leishmania tarentolae (LTE) system. Human proteins were tagged with EGFP at N terminus, and the viral proteins were tagged with mCherry at C terminus. The protein bands were visualized by fluorescence scanning; viral proteins: M,ORF9c, ORF10, and NSP5; human proteins: ACADM, C20orf4, PMPCA, NDFIP2, PPT1, and ARF6.
(B) A plot of representative signals of AlphaLISA interaction assay for VIP/viral protein pairs shown in (A). Zika virus self-dimerizing C-protein tagged with Cherry and EGFP was used as positive interaction control. As the negative control, we used FKBP-rapamycin-binding (FRB) domain.
(C) Graphic summary of the VIPs/SARS-CoV-2 interaction analysis: the confirmed interactions are shown with green circle, whereas interactions that could not be conformed using this assay are depicted as red diamond.
Related to STAR Methods and Figure S6.
Selected CoV-VIPs are enriched for antiviral and proviral factors
To further clarify that a viral epidemic caused the strong burst of selection, and not another ecological pressure acting on the same set of genes, we test whether the 42 selected CoV-VIPs are enriched for genes with antiviral or proviral effects relative to other CoV-VIPs (i.e., loci that are known to have a detrimental or beneficial effect on the virus, respectively). Because the relevant literature for coronaviruses is currently limited, we extend our set of anti- and proviral loci to include loci reported for diverse viruses with high confidence from the general virology literature (STAR Methods; Data S1K and S1L). We find that 21 (50%) of the 42 CoV-VIPs that came under selection ∼900 generations ago have high-confidence anti- or proviral effects (versus 29% for all 420 CoV-VIPs), a significant inflation in such effects (hypergeometric test p = 6.10−4) that further supports our claim that the underlying selective pressure was most likely a viral epidemic.
Selected mutations lie near regulatory variants active in SARS-CoV-2-affected tissues
Coronavirus infections in humans are known to have pathological consequences for specific tissues; therefore, we investigate whether the genes selected in East Asia are also enriched for regulatory functions in similar tissues. In light of our finding that many putative causal mutations in CoV-VIPs are proximal to eQTLs, we investigate whether selected mutations are situated closer to eQTLs for a given tissue than expected by chance, as this would indicate that the tissue was negatively impacted by the virus (prompting the adaptive response). Note that the GTEx eQTLs we use are not specific to a single tissue and are often shared between tissues. However, each tissue still has its own specific combination of eQTLs. Briefly, we estimate a proximity-based metric that quantifies the distance between the location of the causal mutation estimated by iSAFE and the tissue-specific eQTLs for the 42 loci with selection starting ∼900 generations ago and compare this to the same distances observed among randomly sampled sets of CoV-VIPs (Figure 7; STAR Methods).

Figure 7. Proximity of selection signals to GTEx eQTLs at the 42 selected CoV-VIPs compared to random CoV-VIPs
The histogram shows how close selection signals localized by iSAFE peaks are to the GTEx eQTLs from 25 different tissues, at peak-VIPs compared to randomly chosen CoV-VIPs (STAR Methods). How close iSAFE peaks are to GTEx eQTLs compared to random CoV-VIPs is estimated through a proximity ratio. The proximity ratio is described in the STAR Methods. It quantifies how much closer iSAFE peaks are to eQTLs of a specific GTEx tissue, compared to random expectations that take the number and structure of iSAFE peaks as well as the number and structure of GTEx eQTLs into account (STAR Methods). ∗∗∗∗Proximity ratio test p < 0.0001. ∗∗∗Proximity ratio test p < 0.001. ∗∗p < 0.01. ∗p < 0.05. Note that lower proximity ratios can be associated with smaller p values for tissues with more eQTLs (due to decreased null variance; for example, skeletal muscle versus pancreas). Related to STAR Methods and Figure S5.
We find that GTEx lung eQTLs lie closer to predicted causal mutations among the 42 putative selected loci than for any other tissue (p = 3.10−5; Figure 7). Several additional tissues known to be negatively affected by coronavirus—blood and arteries,43,44 adipose tissue,45 and the digestive tract46—also exhibit closer proximities between putative causal loci and eQTLs than expected by chance (Figure 7). Interestingly, the spleen shows no tendency for eQTLs to lie closer to selected loci than expected around 900 generations ago compared to other evolutionary times, perhaps because the spleen is replete with multiple immune cell types that might be more prone to regular adaptation to diverse pathogens over time.47 Note that tissues with more eQTLs tend to have more significant p values. For example, skeletal muscle has a lower proximity ratio than stomach but also a lower p value due to higher statistical power (more eQTLs). However, we find no correlation (Pearson’s correlation test p = 0.6) between the total number of GTEx v8 eQTLs32 for a given tissue and the proximity ratio for each tissue. Thus, different proximity ratios between tissues do not just reflect a statistical power bias. We further show that iSAFE locates adaptation particularly closer to more lung-specific eQTLs compared to other tissues (Figure S7; STAR Methods). Our results indicate that the tissues impacted in the inferred viral epidemic in East Asia match those affected by SARS-CoV-2.
Coronavirus VIPs are enriched for SARS-CoV-2 susceptibility and COVID-19 severity loci
Our results indicate that many of the selected CoV-VIPs now sit at intermediate frequencies in modern East Asian populations. We anticipate that these segregating loci should make a measurable contribution to the inter-individual variation in SARS-CoV-2 susceptibility and COVID-19 severity among contemporary populations in East Asia. While a genome-wide association study (GWAS) scan has yet to be reported for a large East Asian cohort, two GWASs were recently released that used sizable British cohorts to investigate SARS-CoV-2 susceptibility (1,454 cases and 7,032 controls; henceforth called the susceptibility GWAS) and severity (325 cases [deaths] versus 1,129 positive controls; henceforth called the severity GWAS; data from the UK Biobank;22,23 https://grasp.nhlbi.nih.gov/Covid19GWASResults.aspx). Because we use a non-East Asian population, we only ask, as a functional validation of a viral pressure, whether there is an overlap between the selected loci in East Asia and stronger COVID-19 GWAS hits in the UK Biobank. We do not look at all at the directionality or the size of effects. It is indeed unclear that those would be transposable between populations, given that here we provide evidence that different pathogens may have influenced evolution in different human populations. This also means that we make no claim at all here about any decrease or increase of virus susceptibility in any given human population compared to others. Furthermore, we use the UK-Biobank cohort instead of the complete COVID-19 Host Genetics Initiative meta-GWAS data (https://www.covid19hg.org/),7,8 to avoid population stratification to the best extent possible (a legitimate concern with a trait clearly affected by socioeconomic factors).
While we are unable to precisely identify the causal variants for the selected CoV-VIP genes observed in the ancestors of East Asians—nor would these variants necessarily occur as outliers in a GWAS conducted on the British population—we note that it is possible that other variants in the same CoV-VIP genes may also produce variation in SARS-CoV-2 susceptibility and COVID-19 severity among modern British individuals.
By contrasting variants in CoV-VIPs against those in random sets of genes, we find that variants in CoV-VIPs have significantly lower p values for both the susceptibility GWAS and severity GWAS than expected (simple permutation test p < 10−9 for both GWAS tests; STAR Methods). More importantly, the 42 CoV-VIPs with selection starting ∼900 generations ago have even lower GWAS p values compared to other CoV-VIPs (p = 0.0015 for susceptibility GWAS and p = 0.023 for severity; STAR Methods). This result indicates that the selected genes inferred in our study might contribute to individual variation in COVID-19 etiology in modern human populations in the UK, providing further evidence that a coronavirus or another virus with similar host interactions may have been the selection pressure behind the adaptive response we observe in East Asia. Notably, the strongest GWAS hits identified by the COVID-19 Host Genetics Initiative (listed at https://www.covid19hg.org/publications/) do not overlap with the 42 CoV-VIPs selected in East Asia. The lack of overlap is however not surprising and a result of the design of our analysis (STAR Methods).
Selected CoV-VIP genes include multiple known drug targets
Our analyses suggest that the 42 CoV-VIPs identified as putative targets of an ancient coronavirus (or another virus using similar host interactions) epidemic might play a functional role in SARS-CoV-2 etiology in modern human populations. We find that four of these genes (SMAD3, IMPDH2, PPIB, and GPX1) are targets of eleven drugs currently used or investigated in clinical trials to mitigate COVID-19 symptoms (STAR Methods). While this number is not higher than expected when compared to other CoV-VIPs (hypergeometric test p > 0.05), we note that most of the 42 genes identified here have yet to be the focus of trials. In addition to the four selected CoV-VIP genes targeted by coronavirus-specific drugs, five additional selected CoV-VIPs are targeted by multiple drugs to treat a variety of non-coronavirus pathologies (Data S1). An additional six of the 42 selected CoV-VIPs have been identified by Finan et al.48 as part of the “druggable genome” (Data S1M).
Discussion
We identified a set of 42 CoV-VIPs exhibiting a coordinated adaptive response that likely emerged more than 20,000 years ago (Figure 2). This pattern was unique to East Asian populations (as classified by the 1000 Genomes Project). We show that this selection pressure produced a strong response across the 42 CoV-VIP genes that gradually waned and resulted in the selected loci plateauing at intermediate frequencies. Further, we demonstrate that this adaptive response is likely the outcome of a viral epidemic, as attested by the clustering of putatively selected loci around variants that regulate tissues known to exhibit COVID-19-related pathologies, and the enrichment of variants associated with SARS-CoV-2 susceptibility and severity, as well as anti- and proviral functions, among the 42 CoV-VIP genes selected starting around 900 generations ago.
An important limitation is that some of our analyses rely upon comparative datasets that were generated in contemporary human populations that have different ancestries than the East Asian populations where the selected CoV-VIP genes were detected. In particular, both of the eQTL and GWAS datasets come from large studies that are focused on contemporary populations from Europe and none of the five European populations in our study exhibit the selection signals observed in East Asia. More direct confirmation of the causal role of 42 CoV-VIP genes in COVID-19 etiology will require the appropriate GWAS to be conducted in East Asian populations. The detection of genetic associations among the 42 CoV-VIPs in a GWAS on contemporary East Asians would provide further evidence that one or more coronaviruses, or another virus using similar interactions, comprised the selection pressure that drove the observed adaptive response. Moreover, a high-powered GWAS in East Asian populations would be required to identify the loci that currently impact individual variation in COVID-19 etiology in East Asian individuals. Because of these limitations, and because it would be extremely difficult to control for all the other factors that differ across the world (including socioeconomic factors), our results do not represent evidence for any difference in either increased or decreased genetic susceptibility in any human population.
Insights into ancient viral epidemics from modern human genomes
A particularly salient feature of the adaptive response observed for the 42 CoV-VIPs is that selection appears to be acting continuously over an ∼20,000 years period. The profile of selection in the host East Asian populations is consistent with a new viral pressure that ancestral populations had never experienced previously but that subsequently remained present for a very long period of time. As this manuscript was in the final stages of preparation, the first host-virus interactomes were published for SARS-CoV-1 and MERS-CoV,49 which exhibit an extensive overlap with the SARS-CoV-2 interactome used in the present study.19 This suggests that coronaviruses share a broad set of host proteins that they interact with, which should apply to ancient coronaviruses. These patterns are consistent with one or more coronaviruses driving selection that produced the signals reported here. Still, we cannot exclude that another currently unknown type of viruses might have been responsible, which used the same interactions as coronaviruses with human proteins.
Further validation of the historical trajectories of the causal mutations at selected genes is still needed, including more finely resolved temporal and geographic patterns that could be derived from ancient DNA sampled from across East Asia; however, the requisite ancient samples are currently lacking. Nonetheless, we note the geographic origin of several modern outbreaks of coronaviruses in East Asia point to East Asia being a likely location where these ancient populations came into contact with the virus. Our results suggest that East Asia might have also been a natural range for coronavirus reservoir species during the last 25,000 years.50
Applied evolutionary medicine: Using evolutionary information to combat COVID-19
The net result of the ancient selection patterns on the CoV-VIPs in ancient human populations is the creation of genetic differences among individuals now living in East Asia and between East Asians and populations distributed across the rest of the world. As we demonstrate in this study, this evolutionary genetic information can be exploited by statistical analyses to identify loci potentially involved in the epidemiology of modern diseases—COVID-19 in the present case. Such evolutionary information may ultimately assist in the development of future drugs and therapies by complementing information obtained from more traditional epidemiological and biomedical research. While such studies provide information on a specific gene, the evolutionary approach adopted here leverages evolutionary information in modern genomes to identify candidate genomic regions of interest. This is similar to the information provided by GWAS—i.e., lists of variants or genes that are potentially associated with a particular trait or disease—though we note that the information provided by evolutionary analyses comes with an added understanding about the historical processes that created the underlying population genetic patterns.
The current limitation shared by population genomic approaches, such as GWAS and the evolutionary analyses presented here, is that they identify statistical associations rather than causal links. Further evidence of causal relationships between the CoV-VIPs and COVID-19 etiology could be obtained by examining which viral proteins the selected CoV-VIPs interact with, thus establishing the specific viral functions that are affected.
The ultimate confirmation of causality requires functional validation. It remains to be established whether the genes we have identified in this study might help drug-repurposing efforts and provide a basis for future drug and therapeutic development.
By leveraging the evolutionary information contained in publicly available human genomic datasets, we were able to infer ancient viral epidemics impacting the ancestors of contemporary East Asian populations. Importantly, our evolutionary genomics analyses have identified several new candidate genes that might provide novel drug targets (Data S1). More broadly, our findings highlight the utility of thinking about the possible contribution of evolutionary genomic approaches into standard medical research protocols. Indeed, by revealing the identity of our ancient pathogenic foes, evolutionary genomic methods may ultimately improve our ability to predict—and thus prevent—the epidemics of the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| 1000 Genomes Project – Phase 3 | Auton et al.18 | ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/ |
| VIPs | this manuscript | Data S1 |
| Relate-estimated coalescence rates, allele ages and selection P values for the 1000GP | Speidel et al.20 | https://zenodo.org/record/3234689 |
| GTEx expression | GTEx Consortium32 | https://gtexportal.org/home/datasets |
| Protein-protein interactions (IntAct) | Luisi et al.51 | https://www.ebi.ac.uk/intact |
| The density of conserved segments (PhastCons) | Siepel et al.52 | http://hgdownload.cse.ucsc.edu/goldenPath/hg19/phastCons46way/ |
| The density of regulatory elements | N/A | http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeRegDnaseClustered |
| The recombination rate | Hinch et al.53 | https://www.well.ox.ac.uk/∼anjali/AAmap/ |
| Software and algorithms | ||
| selscan (compute nSL). | Szpiech and Hernandez54 | https://github.com/szpiech/selscan |
| hapbin (compute his) | Maclean et al.55 | https://github.com/evotools/hapbin |
| Gene Set Enrichment Pipeline | Enard and Petrov17 | https://github.com/DavidPierreEnard/Gene_Set_Enrichment_Pipeline |
| Relate | Speidel et al.20 | https://myersgroup.github.io/relate/ |
| CLUES | Stern et al.21 | https://github.com/35ajstern/clues |
| iSAFE | Akbari et al.31 | https://github.com/alek0991/iSAFE |
| Reagents | ||
| NucleoBond Xtra Midi kit for transfection-grade plasmid DNA | Machery-Nagel SCIENTIFIX PTY LTD, AUS | catalog #740410.5 |
| Anti-GFP AlphaLISA Acceptor bead | Perkin Elmer | catalog #AL133M |
| Streptavidin Alphascreen Donor bead | Perkin Elmer | catalog #6760002 |
| OptiPlate-384, White Opaque 384-well Microplate | Perkin Elmer | catalog #6007290 |
| Proxy-Plate-384, White shallow 384-well Microplate | Perkin Elmer | catalog #6008280 |
| Bolt 4 to 12%, Bis-Tris, 1.0 mm Mini Protein Gel, 12-well | Thermofisher scientific | catalog #NW04122BOX |
| NuPAGE sample buffer (4x) | Life Technologies | catalog #NP0007 |
| Prestained Protein Ladder, All blue standard | Biorad | catalog #1610373 |
| NuPAGE MOPS SDS Running Buffer (20X) | Thermofisher scientific | catalog #NP0001 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Enard denard@email.arizona.edu.
Materials availability
This study did not generate new unique reagents. The list of reagents used is provided in the Key resources table.
Data and code availability
The pipeline required to reproduce the analysis, as well as a complete list of VIPs for diverse viruses, are available at https://github.com/DavidPierreEnard/Gene_Set_Enrichment_Pipeline
Experimental model and subject details
All the sources of bioinformatic data used in the analysis are provided in the Key resources table.
Method details
Terminology
For convenience, the 42 CoV-VIPs that we infer to have started coming under selection around 900 generations ago are called peak-VIPs in the STAR Methods.
Coronavirus VIPs
We used a dataset of 5,291 VIPs (Data S1A). Of these, 1,920 of these VIPs are high confidence VIPs identified by low-throughput molecular methods, while the remaining VIPs were identified by diverse high-throughput mass-spectrometry studies. Using VIPs to find the genomic footprints of an ancient epidemic is justified by the fact that VIPs do not just interact with viruses. These interactions are in fact functionally consequential for viruses. The 420 CoV-VIPs are part of a much larger set of VIPs found to interact to date with more than 20 different viruses that infect humans16 (Data S1A). In total, there are currently 5,291 VIPs (Data S1A). Of these, 1,920 high confidence VIPs were annotated manually by curating the virology literature and correspond to VIPs that were identified by low-throughput molecular methods.16 These VIPs were often identified by virologists who hypothesized that the interaction existed in the first place based on previous virology knowledge. The other 3,371 VIPs identified by multiple high-throughput mass spectrometry experiments, such as the one conducted to identify the 332 SARS-CoV-2 VIPs.19
To confirm that VIPs are indeed functionally important for viruses beyond just interacting physically, and represent a viable way of detecting specific viral selective pressures that trigger host adaptation, we verify that VIPs have antiviral or proviral effects on the viral replication cycle on which positive selection can act. More specifically, we need to confirm that VIPs have much more frequent proviral or antiviral effects compared to non-VIPs. To test this, we are currently manually annotating all protein-coding genes in the human genome that were involved in published low-throughput expression perturbation experiments to assess their effects on viruses, and manually curable in PubMed. Such expression perturbation experiments typically include RNAi knock-down experiments or overexpression experiments. These experiments are useful to annotate proviral or antiviral effects. Indeed, decreasing the expression of an antiviral VIP should be beneficial to viral replication, while increasing the expression of an antiviral VIP should be detrimental to the virus. Conversely, decreasing the expression of a proviral VIP should be detrimental to viral replication, while increasing the expression of a proviral VIP should be beneficial. We consider only low-throughput expression perturbation experiments, where the expression of only one candidate gene is perturbed. This excludes high throughput genome-wide RNAi screens known for their high false positive and high false negative rates. Using these criteria, we have so far found that 855, or 66% of 1,300 already annotated low-throughput VIPs have a known antiviral or proviral effect. Of the 2,627 high-throughput VIPs that we already annotated, 426 or 16% have a known antiviral or proviral effect. Of the 3,913 non-VIPs that we already annotated, 171 or 4% have a known antiviral of proviral effect. Although we have not annotated all human protein-coding genes yet, the large numbers already annotated imply that these proportions are very likely to be close to the final proportions when all genes are annotated.
Thus, approximately two-thirds of low-throughput VIPs have known antiviral or proviral effects that were revealed by expression perturbation experiments such as gene knock-down or overexpression. The 16% proportion of high throughput VIPs known to have a clear antiviral or proviral effect is much lower than the two-thirds of low-throughput VIPs with antiviral or proviral effects, but it is important to consider that high-throughput VIPs have not been investigated anywhere near as much as the low-throughput ones. In contrast, only 4% of non-VIPs with no known viral interaction have published antiviral or proviral effects. Both low-throughput and high-throughput VIPs are thus far more often functionally consequential for viruses compared to non-VIPs (simple permutation test p < 10−16 in both cases). Note that because they will dilute the signal rather than create it, a certain amount of random, false-positive high-throughput interactions are expected to be conservative when trying to detect ancient epidemics.
Focusing specifically on the 420 CoV-VIPs, we find that 121 or 28.9% of them already have published antiviral or proviral effects (Data S1K). Of the 332 SARS-CoV-2 VIPs, 83 or 25% of them have antiviral or proviral effects (Data S1K), often independently confirmed in multiple viruses. The SARS-CoV-2 VIPs are thus more than six times more likely to have antiviral or proviral effects than non-VIPs, which supports the high quality of the mass spectrometry screen conducted by Gordon et al.,19 as confirmed by our own validations of interactions (Figure 6). Note that it is unrealistic to expect much higher percentages at SARS-CoV-2 VIPs, given that coronaviruses are only starting to be more thoroughly investigated, and have been much less investigated than other viruses such as HIV or IAV (Influenza Virus).
Validation of selected SARS-CoV-2 CoV-VIPs
We co-express the selected SARS-CoV-2 CoV-VIPs:SARS-CoV-2 protein pairs in Leishmania tarentolae (LTE) cell-free protein expression system and test their interactions using AlphaLISA protein: protein interaction assay. This approach (Figure S6A) was previously used for rapid analysis of intra-viral PPI network of ZIKA virus.42 Two of the 35 selected SARS-CoV-2 CoV-VIPs, UBAP2 and FBN2, are missing from the analysis because they are not available in the DNASU plasmid repository (see below). All proteins were tagged with either EGFP or Cherry fluorescent proteins and with the exception of GCC2 and RTN4 could be detected on SDS-PAGE upon cell-free co-expression (Figures 6A and S6B). These two host proteins have large molecular weights (Data S1) that make proper protein folding challenging, which likely explain detection failure. When the in vitro translation reactions were subjected to AlphaLISA interaction analysis, out of 33 interacting protein pairs 24 were positively confirmed by our assay (Figures 6A–6C and S6B). Of the two negative results for GCC2 and RTN4, only GCC2 is represented in lane 23 of Figure S6B for comparison with the positive results. The 73% (24/33) validation rate is likely to be an underestimation of the actual true interactions in this experimental set due to the limitations of the expression system and also the details of biochemistry of the individual proteins. The obtained results probably contain a significant number of false negatives due to two factors. First, we have demonstrated that LTE cell-free system can produce approximately 70% of human proteins in full length, folded and monodispersed form.56 Therefore, it is likely that at least some of the human proteins have not been expressed in functional form. Furthermore, post-translational modifications and functional states of proteins may modulate their interactions with viral ORFs. For example one of these proteins in this set, RhoA, is post translationally prenylated and is carboxyl methylated in vivo.57,58 Due to the lack of isoprenoid pyrophosphate precursors this modification is likely to be absent in its LTE produced version. Furthermore, its nucleotide bound form (GDP versus GTP) modulates its interaction with many RhoA binding proteins59 and may not be optimal in the current experimental set up. Moreover, protein-focused assay optimization is likely to reveal additional positive interactions in this set.
For gene sequences and generation of Cell-free expression vectors, the DNA sequences of SARS-CoV-2 were sourced from the isolate of 2019-nCoV/USA-WA1/2020, (accession number MN985325) and based on the published annotation of the genome sequence of SARS-CoV-2.19 The viral genes were synthesized and inserted into pCellFree_G06 gateway destination vector (available in Addgene, Plasmid # 67140; https://www.addgene.org/67140/ by Gene Universal. The human gene plasmids were generated by DNASU plasmid repository (Arizona State University, US). The genes were cloned into pmCell-free_KA1 gateway destination vector (available in Addgene, Plasmid #145369; https://www.addgene.org/145369/). The synthesized plasmids DNA were amplified and isolated by NucleoBond Xtra Midi kit.
For the Cell-free co-expression of CoV-VIPs and SARS-CoV-2 protein pairs, the protein pairs were co-expressed in the LTE cell-free expression system. The Leishmania tarentolae translation competent extract and the feeding solution for protein expression were prepared as previously described.60 The DNA templates for N-terminal-GFP (8-12 nM) and C-terminal-Cherry (10-15 nM) tagged proteins were added concomitantly to the LTE reaction mixture and the samples were incubated for 5h at 25°C for expression. The expression of proteins was performed using 384-well Proxiplate in 10 μL volume. The Protein expression was detected by measuring GFP and Cherry fluorescence using Tecan Spark multimode microplate reader (Tecan Australia Pty). In addition, for analysis of co-translated eGFP and mCherry fused proteins, the LTE reactions were mixed with 1:1 v/v of 2x NuPAGE sample buffer and loaded on a Bolt 4%–12% Bis-Tris protein gel. The proteins were detected by scanning the gel using ChemiDoc MP System (Bio-Rad, Australia).
AlphaLISA assays were performed in Optiplate-384 plus plates using Anti-GFP AlphaLISA Acceptor and Streptavidin Donor beads. Alpha beads were prepared according to the protocol provided by the manufacturer (https://www.perkinelmer.com/Content/TDLotSheet/AS112D_AS112_2587358.pdf). Briefly, the acceptor and donor beads stocks (5 mg/mL) were diluted to 100 μg/mL (5x) in AlphaLISA assay buffer (Buffer A: 25 mM HEPES, 50 mM NaCl, 0.1% BSA and 0.01% Nonidet P-40; pH:7.5). The biotinylated mCherry nanobody diluted in buffer A (final concentration of 4 nM) was added into microplate wells followed by the addition of lysate containing putative interacting proteins and 5 μL of the acceptor beads (5x). The samples were incubated for 30 min at room temperature. Subsequently, 5 μL of donor beads (5x) were added to samples under low light conditions and incubated for 30 minutes at room temperature. For all experiments, samples were prepared in triplicate and the assay was repeated two times. The AlphaLISA signal was detected with Tecan Spark multimode microplate reader using the following settings: Mode: AlphaLISA, Excitation time: 130 ms, Integration time: 300 ms.
Genomes and sweeps summary statistics
To detect signatures of adaptation in various human populations, we used the 1000 Genomes Project phase 3 dataset18 which provides chromosome level phased data for 26 distinct human populations representing all major continental groups. To measure nSL separately in each of the 26 populations, we use the selscan software available at https://github.com/szpiech/selscan.54 To measure iHS, we use the hapbin software available at https://github.com/evotools/hapbin.55
Ranking of sweep signals at protein-coding genes and varying window sizes
To detect sweep enrichments at CoV-VIPs, we first order, separately in each of the 26 1000 Genomes populations, human Ensembl61 (version 83) protein-coding genes according to the intensity of the sweep signals at each gene. As a proxy for the intensity of these signals, we use the average of either iHS or nSL across all the SNPs with iHS or nSL values within a window of fixed size, centered at the genomic center of genes, halfway between the most upstream transcription start site and the most downstream transcription end site. We then rank the genes according to the average iHS or nSL (more precisely their absolute values) in these windows. We get six rankings for six different fixed window sizes: 50kb, 100kb, 200kb, 500kb, 1,000kb and 2,000kb. We do this to account for the variable size of sweeps of different strengths. We then estimate the sweep enrichment at CoV-VIPs compared to controls over all these different window sizes considered together, or at specific sizes, as described below and in Enard & Petrov.17 Note that we use up to 2,000kb windows to measure the average nSL or iHS, while we use control genes that are at least 500kb away from CoV-VIPs. This means that a fraction of control windows can overlap CoV-VIP windows. This makes our comparison conservative by reducing the visible excess of sweep signals at CoV-VIPs compared to control genes, since a proportion of the controls now also reflect the enrichment at CoV-VIPs, albeit not to the same extent as windows actually centered on CoV-VIPs.
Estimating the whole ranking curve enrichment at CoV-VIPs and its statistical significance
To estimate a sweep enrichment in a set of genes, a typical approach is to use the outlier approach to select, for example, the top 1% of genes with the most extreme signals. Here we use a previously described approach to estimate a sweep enrichment while relaxing the requirement to identify a single top set of genes. Instead of, for example, only estimating an enrichment in the top 100 genes with the strongest sweep signals, we estimate the enrichment over a wide range of top X genes, where X is allowed to vary from the top 10,000 to the top 10 with many intermediate values (10000, 9000, 8000, 7000, 6000, 5000, 4000, 3000, 2500, 2000,1500, 1000, 900, 800, 700, 600, 500, 450, 400, 350, 300, 250, 200, 150, 100, 90, 80, 70, 60, 50, 40, 30, 25, 20, 15, 10). This creates an enrichment curve as in Figure 1. Figure 1 shows the estimated relative fold enrichments at CoV-VIPs compared to controls, from the top 1,000 to the top 10 nSL. The statistical significance of the whole enrichment curve can then be estimated by using block-randomized genomes, as described in Enard & Petrov.17 All the methodological details on how we use block-randomized genomes to estimate the sensitivity and False Positive Risk of the pipeline are described in reference,17 and the readers can refer to that reference for further details. In brief, block-randomized genomes make it possible to generate a large number of random whole enrichment curves while maintaining the same level of clustering of genes in the same candidate sweeps as in the real genome, which effectively controls for gene clustering. Comparing the real whole enrichment curve to the random ones then makes it possible to estimate an unbiased false-positive risk28 (also known as False Discovery Rate in the context of multiple testing) for the observed whole enrichment curve at CoV-VIPs. A single false positive risk can be estimated for not just one curve but by summing over multiple curves combined, thus making it possible to estimate a single false positive risk over any arbitrary numbers of rank thresholds, window sizes, summary statistics, and populations. For instance, we estimate the false-positive enrichment risk of p = 2.10−4 at CoV-VIPs for rank threshold from the top 10,000 to top 10, over six window sizes, for the five East Asian populations in the 1000 Genomes data, and for both nSL and iHS, all considered together at once. This makes our approach more versatile and sensitive to selection signals ranging from a few very strong sweeps, to many, more moderately polygenic hitchhiking signals. The entire pipeline to estimate false-positive risks with block-randomized genomes is available at https://github.com/DavidPierreEnard/Gene_Set_Enrichment_Pipeline. Note that the false positive risk estimates fully take into account the extra variance expected from shrinking the pool of potential control genes by requiring control genes that match CoV-ViPs for multiple confounding factors.17
Building sets of controls matching for confounding factors
To estimate a sweep enrichment at CoV-VIPs, we compare CoV-VIPs with random control sets of genes selected far enough (> 500kb) from CoV-VIPs that they are unlikely to overlap the same large sweeps. We do not compare CoV-VIPs with completely random sets of control genes. Instead, we use a previously described bootstrap test to build random control sets of genes that match CoV-VIPs for a number of potential confounding factors that might explain a sweep enrichment, rather than interactions with viruses. The bootstrap test has been described in detail,17 and is available at https://github.com/DavidPierreEnard/Gene_Set_Enrichment_Pipeline.
We include 11 different potential confounding factors in the bootstrap test:
- •
average GTEx expression in 53 GTEx V8 tissues.
- •
GTEx expression in lymphocytes.
- •
GTEx expression in testis.
- •
the number of protein-protein interactions from the Intact database, curated by Luisi et al.51
- •
the Ensembl (v83) coding sequence density in a 50kb window centered on each gene.
- •
the density of conserved segments identified by PhastCons.52 (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/phastCons46way/).
- •
the density of regulatory elements, estimated by the density of Encode DNase I V3 Clusters (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeRegDnaseClustered/) in a 50kb window centered on each gene.
- •
the recombination rate in a 200kb window centered on each gene.53
- •
the GC content in a 50kb window centered on each gene.
- •
the number of bacteria each gene interacts with, according to the Intact database (as of June 2019; https://www.ebi.ac.uk/intact/).
- •
the proportion of genes that are immune genes according to Gene Ontology annotations GO:0006952 (defense response), GO:0006955 (immune response), and GO:0002376 (immune system process) as of May 2020.
Two other factors commonly controlled for in selection analyses are gene length and SNP density. In our controls, gene length is accounted for by the functional density controls such as the density of coding, conserved, and regulatory elements. Gene length could be an issue if longer genes mean higher densities of functional elements more likely to adapt. But we match functional densities, and thus gene length is not an issue. SNP density could be problematic, because the values of haplotype-based summary statistics such as iHS or nSL can be sensitive to the local SNP density. To test the potential impact of SNP density, we add the number of SNPs in East Asia in 50kb windows centered on genes, and the number of SNPs in larger, 500kb windows centered on genes, to the 11 confounding factors already included in the matching process. We find that adding SNP density to the other confounding factors affects the observed sweep enrichment at CoV-VIPs in East Asia very weakly (top 1,000 rank thresholds, 1Mb and 2Mb windows, nSL+iHS: FPR = 6.10−5; compare Figures S2O–S2S to Figure 1).
We further show that the strong sweep excess at CoV-VIPs is also visible when not controlling for confounding factors at all (Figures S2J–S2N; iHS+nSL FPR < 10−5). This confirms that the control genes selected by the bootstrap test when matching confounding factors are not unusual with respect to their sweep prevalence, as also shown by the FPR analysis.
Host intrinsic functions do not explain the pattern and timing of adaptation at CoV-VIPs
An important limitation to consider when inferring ancient epidemics is that VIPs do not just interact with viruses, but are also involved in multiple hosts intrinsic functions. These host functions could in theory explain the enrichment and timing of adaptation at CoV-VIPs, rather than interactions with a coronavirus or related virus. This would happen as a result of specific host functions being enriched at CoV-VIPs, and also intrinsically enriched in adaptive signals independently of any interaction with viruses. Host functions not enriched at CoV-VIPs are not expected to generate an enrichment in adaptation at CoV-VIPs, because the lack of enrichment means that they are present in similar or smaller proportions in the rest of the genome.
Thus, if host functions enriched at CoV-VIPs, rather than viral interactions, explain adaptation at CoV-VIPs, we expect that i) genes with these host functions should be enriched in sweep signals even when they don’t interact with coronaviruses and ii) genes with these host functions should have started adapting around 900 generations ago, to also explain the timing of adaptation at CoV-VIPs, even when they do not interact with coronaviruses.
To estimate the role of host intrinsic functions, we use the functional annotations from the Gene Ontology (GO) for GO biological processes, GO molecular functions, and GO cellular localizations. In total, there are 106 GO annotations that are enriched at CoV-VIPs compared to the matched controls already used to assess the sweep enrichment (p < 0.001 based on 10,000 matched control sets). Of these 106 GO annotations, only 20 have a more than two-fold enrichment among CoV-VIPs (and 50 genes or more among non-CoV-VIPs; Data S1E) and are thus more likely to contribute to the strong sweep enrichment at CoV-VIPs. We first test if these 20 GO annotations are enriched in sweeps independently of any interaction with coronaviruses. To do this, we use the same bootstrap test used to compare CoV-VIPs with matched controls, but this time we compare genes with the GO annotations, with control genes far enough (> 500kb) from any other gene with these annotations. To make sure that a significant enrichment would have nothing to do with coronaviruses, we exclude from this comparison any gene closer than 500kb to any CoV-VIP. In total, there are 1723 genes with at least one of the 20 highly enriched GO annotations, and 3701 far enough potential control genes. Using exactly the same iHS and nSL enrichment curves used to detect a sweep enrichment at CoV-VIPs (STAR Methods), we do not find any significant enrichment at the 1723 genes compared to matched controls (whole enrichment curves for nSL and iHS combined, p = 0.15). Furthermore, we do not find any significant enrichment in strong sweeps signals within the nSL or iHS top 1000 (p = 0.77), as we do at CoV-VIPs (Figure 1). When considered individually rather than all together, only four of the 20 functions have a significant sweep enrichment (p < 0.05; Data S1E).
To test whether these four functions explain the sweep enrichment at CoV-VIPs, we test this sweep enrichment at CoV-VIPs again, but this time excluding all genes included in the four previous GO annotations. The sweep enrichment at the remaining CoV-VIPs (91% of them) is the same as when testing all CoV-VIPs (the sum of differences between observed and expected numbers over all the nSL sweep rank thresholds, over all window sizes, and over all five East Asian populations is 14,620 for 352 CoV-VIPs included in the test, versus 15,848 for 385 CoV-VIPs included when not excluding GO functions, in other words almost perfectly proportional to the number of genes included in the test), thus showing that these four host functions do not explain the sweep enrichment at CoV-VIPs. Moreover, further excluding all genes with the 20 GO annotations over-represented more than twofold at CoV-VIPs, we find that the remaining CoV-VIPs (58% of them) have a stronger sweep enrichment than when considering all CoV-VIPs (the sum of differences between observed and expected numbers is 10,843 for 222 genes included in this test, proportionally more than the 15,848 sum of differences for 385 CoV-VIPs when not excluding GO functions). Excluding the genes with any of 106 over-represented GO annotations at CoV-VIPs, we also find that the remaining CoV-VIPs (16% of them) have a stronger sweep enrichment than when considering all CoV-VIPs (sum of differences 4,575 for 62 CoV-VIPs). Host intrinsic functions, as annotated by GO, thus cannot explain the sweep enrichment at CoV-VIPs.
Nevertheless, we further test which GO annotations enriched at CoV-VIPs have a significant peak of Relate times around 900 generations ago, as we did before for CoV-VIPs. To do this, we consider all GO annotations enriched at COV-VIPs, but this time compared to completely random controls, rather than compared to control sets matched for confounding factors as before. Indeed, we previously tested the significance of the peak around 900 generations ago at CoV-VIPs compared to completely random controls (STAR Methods), and here we do the same for a fair comparison. Compared to fully random controls, CoV-VIPs are significantly enriched in 316 GO annotations (p < 0.001). Of these 316 GO annotations, 238 are enriched more than two-fold, many more than the 20 GO annotations enriched more than two-fold when using controls matched for confounding factors. This shows that controlling for the confounding factors that we take into account (STAR Methods) effectively controls for many other correlated host intrinsic functions. A total of 39 GO annotations are enriched more than four fold at CoV-VIPs when compared to fully random controls. When considered all together, all the 1,134 genes in the genome other than CoV-VIPs, but with at least one of these 39 highly enriched GO annotations, do not have a significant peak of Relate times (peak significance test p = 0.18). When considered individually, only 16 of all the initial 316 over-represented GO annotations have a significant peak between 770 and 970 generations ago (Data S1F; peak significance test p < 0.05). When removing all CoV-VIPs with at least one of these 16 GO annotations (31% of them), the magnitude of the peak around 900 generations ago at the remaining CoV-VIPs compared to all CoV-VIPs is not affected (Figures S1A and S1B).
Taken together, these results make it very unlikely that host intrinsic functions explain the patterns and timing of adaptation observed at CoV-VIPs, and make a causal role of coronavirus-like viruses more plausible. Below, we provide further, virus-focused functional evidence, further supporting this.
Estimating adaptation start times at specific genes with Relate
As times of emergence of adaptive mutations, we use the publicly available estimates from Relate (https://myersgroup.github.io/relate/). Relate estimates mutation emergence times while controlling for fluctuations of population size over time, based on the coalescence rates it reconstructs after inferring ancestral recombination graphs at the scale of the whole genome.20 Relate provides two times of emergence of mutations, one low estimate (less generations ago), and one high estimate (more generations ago). The low time estimate corresponds to the time when Relate estimates an elevated probability that the frequency of the mutation is different from zero (95% confidence interval, most recent time estimate). The high time estimate corresponds to the time when Relate estimates that the probability is not too small that the frequency of the mutation is different from zero. For our purpose of estimating when selection started, the low time estimate is the best suited, because it provides an estimate of when the frequency of a selected mutation was already high enough to distinguish from zero, for those mutations where selection started from a very low frequency. For cases where selection started with standing genetic variants that were already distinguishable from zero, the Relate low time estimates for the emergence of mutations do not provide a good proxy for when selection actually started. Thus, if we were able to estimate when selection started for standing genetic variants, we might be able to observe an even stronger peak than the one we see when just relying on those variants where selection started from low frequencies.
Using the low Relate time estimates is also justified due to the fact that the sweep establishment phase can take very variable amounts of time before the start of the sweep exponential phase. During the establishment phase, selected alleles are still mostly governed by drift which makes pinpointing the actual starting time of selection difficult. In this context, the low Relate time estimates provide an estimate of the time when the selected alleles were no longer at very low frequencies not statistically different from zero, and closer to entering the exponential phase, which provides a more certain time estimate for when selection started for certain.
An important step is then to choose at each CoV-VIP locus, and all the other control loci, which Relate mutation to use to get a single time estimate for each locus. Note that here we make an assumption that each locus has experienced only one single adaptive event. Given our finding that iSAFE peaks at CoV-VIPs are much closer to GTEx V8 eQTLs than expected by chance, it is likely that the selected adaptive mutations are regulatory mutations at, or close to annotated eQTLs for a specific gene. They are not necessarily exactly located at eQTLs, because current eQTLs annotations may still be incomplete, and in our case we use eQTLs identified in GTEx V8 using mostly European individuals, even though we analyze selection signals in East Asian populations. Because of these limitations, we use the Relate estimated time at the mutation where Relate estimates the lowest positive selection p value within 50kb windows centered on eQTLs. We also only consider variants with a minor allele frequency greater than 20%, given the signals detected by iHS and nSL that only have some power to detect incomplete sweeps above 20% frequencies.24,25 This also excludes a potential risk of confounding by low frequency neutral or weakly deleterious variants, that can show selection-like patterns when their only way to escape removal early on is through a chance, rapid frequency increase that can look like selection. The Relate selection test is based on faster than expected coalescence rates given the population size at any given time, and its results are publicly available at https://myersgroup.github.io/relate/. Note that the mutation with the lowest Relate p value does not always overlap with an iSAFE peak (Figure S5A), which is not entirely surprising if the haplotype signals exploited by both Relate and iSAFE partly deteriorated due to recombination since the time selection at CoV-VIPs was strong (Figures 3 and 5). Both of these methods are indeed designed to locate the selected variant right after, or during, active selection.
Because we work with five different East Asian populations, we more specifically select the variant with the lowest Relate selection test p value on average across all the five East Asian populations. Then, we also use the corresponding average low Relate mutation time estimate across the five East Asian populations. We do not attempt to estimate the selection time and p value by considering all 1000 Genomes East Asian individuals tested together by Relate, because then the Relate selection test is at a greater risk of being confounded by population structure. Finally, we only consider CoV-VIPs and other control genes with an average Relate selection test p value lower than 10−3, to make sure that we indeed use estimated times at selected variants.
The peak significance test
To test if the peak of Relate time estimates around 900 generations ago at CoV-VIPs (Figure 2) is expected simply by chance or not, we designed a peak significance test. The test compares the peak at CoV-VIPs, with the top peaks obtained when repeatedly randomly sampling sets of genes. We first identify the most prominent peak at CoV-VIPs by visual inspection of the pink distribution of Relate times for CoV-VIPs compared to the blue distribution of Relate times for all protein-coding genes with an estimated Relate time (Figure 2). To build these distributions, top Relate selected mutations shared between multiple neighboring genes (CoV-VIPs or controls) are counted only once, to avoid a confounding effect of gene clustering (152 selected variants at CoV-VIPs, 1771 selected variants for all protein coding genes). The peak around 900 generations ago (870 generations more exactly) spans approximately 200 generations, where the pink distribution is clearly above the blue one. We then use a 200 generations-wide window, sliding every generation from 0 to 6,000 generations to verify the peak more rigorously. Sliding one generation after another, each time we count the difference between the number of Relate selected variants at CoV-VIPs that fall in the sliding 200 generations window, and the number of Relate selected variants at all other genes that are not CoV-VIPs, weighted by the percentage of variants found at CoV-VIPs, to correct for the different size of the two sets of variants. Using this sliding window approach, the top of the peak is found at 870 generations, with a difference of 19.5 additional Relate selected variants between 770 and 970 (870 plus or minus 100) at CoV-VIPs compared to the null expectation.
We then repeat the sliding of a 200 generations window to identify the maximum peak and measure the same difference, but this time for random sets of Relate selected variants of the same size (152 selected variants out of the 1,771 selected variants). To estimate p values, we then compare the actual observed difference with the distribution of differences generated with one million random samples.
As mentioned in the Results, one potential issue is that we run the peak significance test after we already know that CoV-VIPs are enriched for iHS and nSL top sweeps, and especially enriched for nSL top sweeps. This enrichment may skew the null expectation for the distribution of Relate times at CoV-VIPs. In other words, there is a risk that any set of genes with the same sweep enrichment might exhibit the same peak as CoV-VIP. As a result, comparing CoV-VIPs with randomly chosen non-CoV-VIPs may not be appropriate. To test this, we repeat the peak significance test, but this time comparing the peak at CoV-VIPs with the peaks at random sets of non-CoV-VIPs that we build to have the same distribution of nSL ranks as CoV-VIPs. To do this, we define nSL bins between ranks 1 and the highest rank with a rank step of 100 between each bin, and we count how many Relate selected variants fall in each bin (each gene has one nSL rank and one Relate selected variant). To build the random set, we then fill each of the 100 bins with the same number of random non-CoV-VIPs, as long as their nSL rank falls within that bin. We use the average nSL rank over the five East Asian populations, and the lower population-averaged rank of either 1 Mb or 2Mb window sizes (where we observe the strongest enrichment at CoV-VIPs, see Results). The results of the peak significance test are unchanged when using the matching nSL distribution (peak significance test p = 1.10-4 versus p = 2.3.10-4 without matching nSL distribution).
In further agreement with the fact that the sweep enrichment does not confound the peak significance test, the peak at CoV-VIPs stands out more when repeating the peak significance test using a smaller nSL top rank limit (Figure S1C). In this case, we compare sets of CoV-VIPs and sets of controls both enriched in stronger sweep signals. Thus, if stronger sweep signals at CoV-VIPs biased the peak significance test, we would expect the peak to fade away when comparing only CoV-VIPs and controls both with stronger nSL signals. Conversely, we observe that half of the CoV-VIPs with the weaker nSL signals (population-averaged nSL rank higher than 7,200 for both 1Mb and 2Mb windows) do not show a significant peak (peak significance test p = 0.53).
The iSAFE peaks/eQTL proximity test
Adaptation in the human genome was likely mostly regulatory adaptation through gene expression changes.26,33, 34, 35 To test if positive selection at CoV-VIPs involved regulatory changes, we ask whether the signals of adaptation around CoV-VIPs are localized closer than expected by chance to GTEx eQTLs that affect the expression of CoV-VIPs in present human populations. We use proximity instead of exact colocalization because we do not expect selection signals in East Asia to colocalize perfectly with eQTLs identified mostly from European tissue samples. The genomic regions at or close to CoV-VIP GTEx eQTLs are likely enriched for CoV-VIP regulatory elements, and therefore the most likely place to find CoV-VIP-related adaptations in the genome. To localize where adaptation occurred, we use the iSAFE method that was specifically designed for this purpose.31 iSAFE scans the genome and estimates a score that increases together with proximity to the actual selected mutation. The higher the score, the higher the odds that the scored variant is itself the selected one, or close to the selected one. An important caveat is that iSAFE is designed to localize where selection happened right after it happened, or as selection is still ongoing. In our case, we have evidence that selection was strong at CoV-VIPs only more than 500 generations (∼14,000 years) ago, and then much weaker more recently (Figure 5). This could be an issue, because we expect that recombination events that occurred after the strong selection might have deteriorated the iSAFE signal that relies on haplotype structure. This is because recombination mixes together the haplotypes that hitchhiked with the selected mutation, with those that did not. In line with this, we often do not observe simple, clean iSAFE score peaks, but instead, iSAFE score plateaus and more rugged peaks (Figure S5A). For this reason, we designed an approach to not only identify the top of simple iSAFE peaks, but also more rugged peaks or plateaus. First, to measure iSAFE scores, we combine all the haplotypes from the five East Asian populations together as input, since we found that the selection signal at CoV-VIPs is common to all these populations (iSAFE parameters:–IgnoreGaps–MaxRegionSize 250000000–window 300–step 100–MaxFreq 0.95–MaxRank 15). We then use a 500kb window sliding every 10kb to identify the highest local iSAFE value in the 500kb window (Figure S5B). Once we have the highest local iSAFE value and coordinate, we define a broader iSAFE peak as the region both upstream and downstream where the iSAFE values are still within 80% of the maximum value (Figure S5B). This way, we can better annotate iSAFE plateaus and rugged peaks, and take into account the fact that they can span more than just a narrow local maximum (Figure S5A).
Once the local iSAFE peaks are identified, we can ask how close GTEx eQTLs are to these peaks compared to random expectations. We first measure the distance of each CoV-VIP GTEx eQTL to the closest iSAFE peak. To avoid redundancy, we merge eQTLs closer than 1kb to each other into one test eQTL at the closest, lower multiple of 1,000 genomic coordinates (for example 3,230 and 3,950 would both become 3,000). We then measure the average of the log of the distance between all CoV-VIPs and their closest iSAFE peak. We use the log (base 10) of the distance, because it matters if the eQTL/iSAFE peak distance is 100 bases instead of 200kb, but it does not really matter if the distance is 200kb or 600kb, because the iSAFE peak at 200kb is likely not more related to the eQTL than the peak at 600kb. Once we have the average of log-distances, we compare it to its random expected distribution. To get this random distribution, we measure the log-distance between each CoV-VIP eQTL and the iSAFE peaks, but after shifting the iSAFE scores left or right by a random value between 1Mb and 2.5Mb (Figure S5B; less, or no shift at all if this falls within telomeres or centromeres). We shift by at least 1Mb to make sure that we do not rebuild the original overlap of iSAFE peaks with eQTLs again and again (some iSAFE peaks, or more precisely rugged peaks and plateaus can be wide and include several hundred kilobases; see Figure S5A). The random shifting effectively breaks the relationship between eQTLs and iSAFE peaks, while maintaining the same overall eQTL and peak structure (and thus variance for the test). The random log-distance distribution then provides an overall random average log-distance to compare the observed average long-distance with, as well as estimate a p value.
Then, to more specifically ask if lung eQTLs at CoV-VIPs or the eQTLs of other specific tissues are closer to iSAFE peaks than expected by chance, we can do the same but only using the eQTLs of that specific tissue. The analysis represented in Figure 7 is however more complicated than just testing if CoV-VIP eQTLs for a specific tissue are closer to iSAFE peaks than expected by chance by randomly sliding iSAFE values. Instead, what we ask is whether the 42 peak-VIPs have eQTLs for a given tissue that are even closer to iSAFE peaks than the eQTLs of all CoV-VIPs in general. To test this, for example with lung eQTLs, we first estimate how close lung eQTLs are to iSAFE peaks at peak-VIPs, compared to random expectations, by measuring the difference between the observed and the average random log-distance, just as described before. We then count the number of peak-VIPs with lung eQTLs (19 out of 25 peak-VIPs with GTEx eQTLs), and we randomly select the same number of any CoV-VIP (which may randomly include peak-VIPs) as long as the random set of CoV-VIPs has the same number of lung eQTLs (plus or minus 10%) as the set of peak VIPs with lung eQTLs (the same gene can have multiple eQTLs for one tissue). We make sure that the tested and the random sets have similar numbers of genes and eQTLs so that the test has the appropriate null variance. We then measure the difference between the observed log-distance, and the randomly expected average log-distance for the random set of CoV-VIPs, exactly the same way we did before for the actual set of peak-VIPs. We then measure the ratio of the observed difference in log-distance between peak-VIPs and the random expectation after many random shiftings (1,000), divided by the average of the same difference measured over many random sets of CoV-VIPs. The final ratio tells us how much closer lung eQTLs are to iSAFE peaks at peak-VIPs compared to CoV-VIPs in general, and still takes the specific eQTLs and iSAFE peak structures at each locus into account, since we compare differences in log-distances expected while preserving the same eQTL and iSAFE peak structure (see above the description of the random coordinate shifting). One important last detail about the test is that because we already found that the 50% of loci with the lowest nSL signals do not show a peak of selection at CoV-VIPs around 900 generations ago (see Results), we do not use these loci in this test since any iSAFE peak there is much more likely to represent random noise, not actual selection locations, and thus likely to dilute genuine signals. Using this test, we find that lung and other tissues’ eQTLs at peak-VIPs are much closer to iSAFE peaks than they are at CoV-VIPs in general. This test thus specifically tells that adaptation happened closer to lung eQTLs, specifically around 900 generations ago compared to other evolutionary times. By estimating the same ratio for 24 other tissues with at least 10 peak-VIPs with the specific tested tissue eQTLs, we can finally rank each tissue for its more pronounced involvement in adaptation ∼900 generations ago, as done in Figure 7. It is particularly interesting in this respect that the tissue with least evidence for being more involved in adaptation at that time more than other evolutionary times is spleen. Spleen indeed likely represents a good negative control as a tissue strongly enriched in immune cell types and likely to have evolved adaptively for most of evolution.
A possible limitation is that eQTLs tend to be shared between many tissues, and only a minority of eQTLs are tissue-specific. This means that in our analysis, specific tissues may stand out only because they share their eQTLs with other tissues that were the primary targets of selection. In order to better identify which specific tissues may have been the strongest targets of selection, we consider again the 42 CoV-VIPs selected 900 generations ago, but this time we ask how much closer than expected by chance their eQTLs are to the location of selection (estimated by iSAFE), as a function of increasing eQTL tissue specificity. We define the tissue specificity of a given eQTL for tissue A as the total number of tissues where GTEx found the eQTL (tissue A + other tissues). We find that for most tissues, eQTLs that are increasingly more specific to these tissues, also tend to be found more and more randomly located compared to the location of selection (Figure S7). Out of 25 tissues, lung is the only one with a clear pattern of more lung-specific eQTLs being closer to the location of selection compared to random expectations (Figure S7, red curve).
UK Biobank GWAS analysis
To compare the UK Biobank GWAS p values at different loci, we assigned one p value for each gene, either CoV-VIPs, peak-VIPs or other genes, even though each gene locus can have many variants with associated GWAS p values. To assign just one single GWAS p value to each gene, we selected the variant with the lowest p value at or very close (< 1kb) to GTEx eQTLs for a specific gene, in line with the fact that GWAS hits tend to overlap eQTLs ,62 and to remain consistent with the rest of our manuscript. We then compared the average p value between different sets of genes using classic permutations (one billion iterations).
We note that the top-ranking loci identified by the COVID-19 Host Genetics Initiative (IFNAR2, OAS, RAVER1, DPP9, LZTFL1, etc.) are broadly acting immune factors. These factors do not interact with viral proteins, and are instead involved in immune signaling cascades that are not specific to a given virus. We therefore do not expect an overlap with the more coronavirus-specific CoV-VIPs that we use here. We also note that we do not necessarily expect the strongest GWAS hits in Europe to be strong hits in other populations. This is particularly true when the investigated trait is the response to a pathogen, given that we show in this manuscript that the evolution of this response was probably population-specific. In addition, although adaptation implies a functional genetic effect, a genetic effect does not necessarily mean it has adaptive potential. Finally, the list of the very top GWAS hits might be sensitive to population stratification, and still change depending on how much population stratification is controlled for. The average strength of the GWAS hits over many Cov-VIPs that we focus on is likely to be less sensitive to these issues. The lack of overlap with the strongest COVID-19 Host Genetics Initiative hits is therefore not very surprising. It also does not take away the fact that we found an enrichment in stronger GWAS hits on average at CoV-VIPs and especially at selected CoV-VIPs.
Drug targets identification
We queried the databases DGIdb,63 and PanDrugs64 for drugs targeting CoV-VIPs and peak-VIPs. For hits from PanDrugs we limited the results to only genes that are in direct interaction with the designated drug. Drugs targeting peak-VIPs are presented in Data S1M. In addition, we present a list of peak-VIPs that are not currently drug targets, but have been previously identified in Finan et al.48 as viable drug targets (druggable genome).
Quantification and statistical analysis
The Method details provide in-depth descriptions of the quantifications and statistical analyses used in this manuscript.
Acknowledgments
We wish to thank Leo Speidel and Aaron Stern for their valuable help using Relate and CLUES, respectively. K.A. is supported by Perkin Elmer Australia and by RISE. Y.S. is supported by the Australian Research Council (ARC DP190103705). R.T. is an ARC DECRA fellow (DE190101069). N.J.K. is funded by grants from the NIH (P50AI150476, U19AI135990, U19AI135972, R01AI143292, R01AI120694, P01AI063302, and R01AI122747); by the Excellence in Research Award (ERA) from the Laboratory for Genomics Research (LGR), a collaboration between UCSF, UCB, and GSK (no. 133122P); by a Fast Grant for COVID-19 from the Emergent Ventures program at the Mercatus Center of George Mason University; by the Roddenberry Foundation; and by funding from F. Hoffmann-La Roche and Vir Biotechnology and gifts from QCRG philanthropic donors. For this work, N.J.K. was supported by the Defense Advanced Research Projects Agency (DARPA) under cooperative agreement no. HR0011-19-2-0020. The views, opinions, and/or findings contained in this material are those of the authors and should not be interpreted as representing the official views or policies of the Department of Defense or the U.S. Government.
Author contributions
Conceived and designed the experiments, Y.S., R.T., K.A., and D.E.; performed the experiments, Y.S., M.E.L., R.T., S.V.M., W.A.J., and D.E.; interpreted the results, Y.S., M.E.L., R.T., C.D.H., A.S.J., S.V.M., W.A.J., K.A., and D.E.; wrote the manuscript, Y.S., R.T., S.V.M., and D.E.; contributed resources/reagents, N.J.K., K.A., and D.E.
Declaration of interests
The Krogan Laboratory has received research support from Vir Biotechnology and F. Hoffmann-La Roche. N.J.K. has consulting agreements with the Icahn School of Medicine at Mount Sinai, New York, Maze Therapeutics, and Interline Therapeutics; is a shareholder of Tenaya Therapeutics; and has received stocks from Maze Therapeutics and Interline Therapeutics. The other authors declare no competing interests.
Supplemental information
Document S1. Figures S1–S7.
References
- 1
- X. Ou, Y. Liu, X. Lei, P. Li, D. Mi, L. Ren, L. Guo, R. Guo, T. Chen, J. Hu, et al.Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoVNat. Commun., 11 (2020), p. 1620
- 2
- C. Hoffman, B.S. KampsSARS ReferenceFlying Publisher (2003)
- 3
- E. Dong, H. Du, L. GardnerAn interactive web-based dashboard to track COVID-19 in real timeLancet Infect. Dis., 20 (2020), pp. 533-534
- 4
- O.D. Balogun, V.J. Bea, E. PhillipsDisparities in cancer outcomes due to COVID-19-a tale of 2 citiesJAMA Oncol., 6 (2020), pp. 1531-1532
- 5
- N. Sattar, I.B. McInnes, J.J.V. McMurrayObesity is a risk factor for severe COVID-19 infection: multiple potential mechanismsCirculation, 142 (2020), pp. 4-6
- 6
- C. Scarpone, S.T. Brinkmann, T. Große, D. Sonnenwald, M. Fuchs, B.B. WalkerA multimethod approach for county-scale geospatial analysis of emerging infectious diseases: a cross-sectional case study of COVID-19 incidence in GermanyInt. J. Health Geogr., 19 (2020), p. 32
- 7
- The COVID-19 Host Genetics InitiativeThe COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemicEur. J. Hum. Genet., 28 (2020), pp. 715-718
- 8
- A. Ganna, The COVID-19 Host Genetics InitiativeMapping the human genetic architecture of COVID-19 by worldwide meta-analysismedRxiv (2021), 10.1101/2021.03.10.21252820
- 9
- G.H.L. Roberts, D.S. Park, M.V. Coignet, S.R. McCurdy, S.C. Knight, R. Partha, B. Rhead, M. Zhang, N. Berkowitz, A.S. Team, et al.AncestryDNA COVID-19 host genetic study identifies three novel locimedRxiv (2020), 10.1101/2020.10.06.20205864
- 10
- D. Ellinghaus, F. Degenhardt, L. Bujanda, M. Buti, A. Albillos, P. Invernizzi, J. Fernández, D. Prati, G. Baselli, R. Asselta, et al., Severe Covid-19 GWAS GroupGenomewide association study of severe Covid-19 with respiratory failureN. Engl. J. Med., 383 (2020), pp. 1522-1534
- 11
- H. Zeberg, S. PääboThe major genetic risk factor for severe COVID-19 is inherited from NeanderthalsNature, 587 (2020), pp. 610-612
- 12
- L.B. Barreiro, M. Ben-Ali, H. Quach, G. Laval, E. Patin, J.K. Pickrell, C. Bouchier, M. Tichit, O. Neyrolles, B. Gicquel, et al.Evolutionary dynamics of human Toll-like receptors and their different contributions to host defensePLoS Genet., 5 (2009), p. e1000562
- 13
- D. Enard, L. Cai, C. Gwennap, D.A. PetrovViruses are a dominant driver of protein adaptation in mammalseLife, 5 (2016), p. e12469
- 14
- S.L. Sawyer, L.I. Wu, M. Emerman, H.S. MalikPositive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domainProc. Natl. Acad. Sci. USA, 102 (2005), pp. 2832-2837
- 15
- L.H. Uricchio, D.A. Petrov, D. EnardExploiting selection at linked sites to infer the rate and strength of adaptationNat. Ecol. Evol., 3 (2019), pp. 977-984
- 16
- D. Enard, D.A. PetrovEvidence that RNA viruses drove adaptive introgression between Neanderthals and modern humansCell, 175 (2018), pp. 360-371.e13
- 17
- D. Enard, D.A. PetrovAncient RNA virus epidemics through the lens of recent adaptation in human genomesPhilos. Trans. R. Soc. Lond. B Biol. Sci., 375 (2020), p. 20190575
- 18
- A. Auton, L.D. Brooks, R.M. Durbin, E.P. Garrison, H.M. Kang, J.O. Korbel, J.L. Marchini, S. McCarthy, G.A. McVean, G.R. Abecasis, 1000 Genomes Project ConsortiumA global reference for human genetic variationNature, 526 (2015), pp. 68-74
- 19
- D.E. Gordon, G.M. Jang, M. Bouhaddou, J. Xu, K. Obernier, K.M. White, M.J. O’Meara, V.V. Rezelj, J.Z. Guo, D.L. Swaney, et al.A SARS-CoV-2 protein interaction map reveals targets for drug repurposingNature, 583 (2020), pp. 459-468
- 20
- L. Speidel, M. Forest, S. Shi, S.R. MyersA method for genome-wide genealogy estimation for thousands of samplesNat. Genet., 51 (2019), pp. 1321-1329
- 21
- A.J. Stern, P.R. Wilton, R. NielsenAn approximate full-likelihood method for inferring selection and allele frequency trajectories from DNA sequence dataPLoS Genet., 15 (2019), p. e1008384
- 22
- C. Bycroft, C. Freeman, D. Petkova, G. Band, L.T. Elliott, K. Sharp, A. Motyer, D. Vukcevic, O. Delaneau, J. O’Connell, et al.The UK Biobank resource with deep phenotyping and genomic dataNature, 562 (2018), pp. 203-209
- 23
- C. Sudlow, J. Gallacher, N. Allen, V. Beral, P. Burton, J. Danesh, P. Downey, P. Elliott, J. Green, M. Landray, et al.UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old agePLoS Med., 12 (2015), p. e1001779
- 24
- A. Ferrer-Admetlla, M. Liang, T. Korneliussen, R. NielsenOn detecting incomplete soft or hard selective sweeps using haplotype structureMol. Biol. Evol., 31 (2014), pp. 1275-1291
- 25
- B.F. Voight, S. Kudaravalli, X. Wen, J.K. PritchardA map of recent positive selection in the human genomePLoS Biol., 4 (2006), p. e72
- 26
- D. Enard, P.W. Messer, D.A. PetrovGenome-wide signals of positive selection in human evolutionGenome Res., 24 (2014), pp. 885-895
- 27
- D.R. SchriderBackground selection does not mimic the patterns of genetic diversity produced by selective sweepsGenetics, 216 (2020), pp. 499-519
- 28
- D. ColquhounThe false positive risk: a proposal concerning what to do about p-valuesAm. Stat., 73 (2019), pp. 192-201
- 29
- Gene Ontology ConsortiumGene Ontology Consortium: going forwardNucleic Acids Res., 43 (2015), pp. D1049-D1056
- 30
- P.C. Sabeti, S.F. Schaffner, B. Fry, J. Lohmueller, P. Varilly, O. Shamovsky, A. Palma, T.S. Mikkelsen, D. Altshuler, E.S. LanderPositive natural selection in the human lineageScience, 312 (2006), pp. 1614-1620
- 31
- A. Akbari, J.J. Vitti, A. Iranmehr, M. Bakhtiari, P.C. Sabeti, S. Mirarab, V. BafnaIdentifying the favored mutation in a positive selective sweepNat. Methods, 15 (2018), pp. 279-282
- 32
- GTEx ConsortiumThe GTEx Consortium atlas of genetic regulatory effects across human tissuesScience, 369 (2020), pp. 1318-1330
- 33
- S. Kudaravalli, J.B. Veyrieras, B.E. Stranger, E.T. Dermitzakis, J.K. PritchardGene expression levels are a target of recent natural selection in the human genomeMol. Biol. Evol., 26 (2009), pp. 649-658
- 34
- Y. Nédélec, J. Sanz, G. Baharian, Z.A. Szpiech, A. Pacis, A. Dumaine, J.C. Grenier, A. Freiman, A.J. Sams, S. Hebert, et al.Genetic ancestry and natural selection drive population differences in immune responses to pathogensCell, 167 (2016), pp. 657-669.e21
- 35
- H. Quach, M. Rotival, J. Pothlichet, Y.E. Loh, M. Dannemann, N. Zidane, G. Laval, E. Patin, C. Harmant, M. Lopez, et al.Genetic adaptation and Neandertal admixture shaped the immune system of human populationsCell, 167 (2016), pp. 643-656.e17
- 36
- P. Moorjani, S. Sankararaman, Q. Fu, M. Przeworski, N. Patterson, D. ReichA genetic method for dating ancient genomes provides a direct estimate of human generation interval in the last 45,000 yearsProc. Natl. Acad. Sci. USA, 113 (2016), pp. 5652-5657
- 37
- M. Ghafari, P. Simmonds, O.G. Pybus, A. KatzourakisPrisoner of War dynamics explains the time-dependent pattern of substitution rates in virusesbioRxiv (2021), 10.1101/2021.02.09.430479
- 38
- L.K. Hayward, G. SellaPolygenic adaptation after a sudden change in environmentbioRxiv (2019), 10.1101/792952
- 39
- S.M. Best, P.J. KerrCoevolution of host and virus: the pathogenesis of virulent and attenuated strains of myxoma virus in resistant and susceptible European rabbitsVirology, 267 (2000), pp. 36-48
- 40
- A.J. Stern, L. Speidel, N.A. Zaitlen, R. NielsenDisentangling selection on genetically correlated polygenic traits via whole-genome genealogiesAm. J. Hum. Genet., 108 (2021), pp. 219-239
- 41
- D. Mellacheruvu, Z. Wright, A.L. Couzens, J.P. Lambert, N.A. St-Denis, T. Li, Y.V. Miteva, S. Hauri, M.E. Sardiu, T.Y. Low, et al.The CRAPome: a contaminant repository for affinity purification-mass spectrometry dataNat. Methods, 10 (2013), pp. 730-736
- 42
- S. Varasteh Moradi, D. Gagoski, S. Mureev, P. Walden, K.A. McMahon, R.G. Parton, W.A. Johnston, K. AlexandrovMapping interactions among cell-free expressed Zika virus proteinsJ. Proteome Res., 19 (2020), pp. 1522-1532
- 43
- C. Bao, X. Tao, W. Cui, B. Yi, T. Pan, K.H. Young, W. QianSARS-CoV-2 induced thrombocytopenia as an important biomarker significantly correlated with abnormal coagulation function, increased intravascular blood clot risk and mortality in COVID-19 patientsExp. Hematol. Oncol., 9 (2020), p. 16
- 44
- C. Grosse, A. Grosse, H.J.F. Salzer, M.W. Dünser, R. Motz, R. LangerAnalysis of cardiopulmonary findings in COVID-19 fatalities: high incidence of pulmonary artery thrombi and acute suppurative bronchopneumoniaCardiovasc. Pathol., 49 (2020), p. 107263
- 45
- K. Michalakis, I. IliasSARS-CoV-2 infection and obesity: common inflammatory and metabolic aspectsDiabetes Metab. Syndr., 14 (2020), pp. 469-471
- 46
- B.J. Elmunzer, R.L. Spitzer, L.D. Foster, A.A. Merchant, E.F. Howard, V.A. Patel, M.K. West, E. Qayed, R. Nustas, A. Zakaria, et al., North American Alliance for the Study of Digestive Manifestations of COVID-19Digestive manifestations in patients hospitalized with coronavirus disease 2019Clin. Gastroenterol. Hepatol. (2020), 10.1016/j.cgh.2020.09.041Published online October 1, 2020
- 47
- L. Quintana-MurciHuman immunology through the lens of evolutionary geneticsCell, 177 (2019), pp. 184-199
- 48
- C. Finan, A. Gaulton, F.A. Kruger, R.T. Lumbers, T. Shah, J. Engmann, L. Galver, R. Kelley, A. Karlsson, R. Santos, et al.The druggable genome and support for target identification and validation in drug developmentSci. Transl. Med., 9 (2017), p. eaag1166
- 49
- D.E. Gordon, J. Hiatt, M. Bouhaddou, V.V. Rezelj, S. Ulferts, H. Braberg, A.S. Jureka, K. Obernier, J.Z. Guo, J. Batra, et al., QCRG Structural Biology Consortium, Zoonomia ConsortiumComparative host-coronavirus protein interaction networks reveal pan-viral disease mechanismsScience, 370 (2020), p. eabe9403
- 50
- A.C.P. Wong, X. Li, S.K.P. Lau, P.C.Y. WooGlobal epidemiology of bat coronavirusesViruses, 11 (2019), p. E174
- 51
- P. Luisi, D. Alvarez-Ponce, M. Pybus, M.A. Fares, J. Bertranpetit, H. LaayouniRecent positive selection has acted on genes encoding proteins with more interactions within the whole human interactomeGenome Biol. Evol., 7 (2015), pp. 1141-1154
- 52
- A. Siepel, G. Bejerano, J.S. Pedersen, A.S. Hinrichs, M. Hou, K. Rosenbloom, H. Clawson, J. Spieth, L.W. Hillier, S. Richards, et al.Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomesGenome Res., 15 (2005), pp. 1034-1050
- 53
- A.G. Hinch, A. Tandon, N. Patterson, Y. Song, N. Rohland, C.D. Palmer, G.K. Chen, K. Wang, S.G. Buxbaum, E.L. Akylbekova, et al.The landscape of recombination in African AmericansNature, 476 (2011), pp. 170-175
- 54
- Z.A. Szpiech, R.D. Hernandezselscan: an efficient multithreaded program to perform EHH-based scans for positive selectionMol. Biol. Evol., 31 (2014), pp. 2824-2827
- 55
- C.A. Maclean, N.P. Chue Hong, J.G. Prendergasthapbin: an efficient program for performing haplotype-based scans for positive selection in large genomic datasetsMol. Biol. Evol., 32 (2015), pp. 3027-3029
- 56
- D. Gagoski, M.E. Polinkovsky, S. Mureev, A. Kunert, W. Johnston, Y. Gambin, K. AlexandrovPerformance benchmarking of four cell-free protein expression systemsBiotechnol. Bioeng., 113 (2016), pp. 292-300
- 57
- P.S. Backlund Jr.Post-translational processing of RhoA. Carboxyl methylation of the carboxyl-terminal prenylcysteine increases the half-life of RhoaJ. Biol. Chem., 272 (1997), pp. 33175-33180
- 58
- I. Cushman, P.J. CaseyRHO methylation matters: a role for isoprenylcysteine carboxylmethyltransferase in cell migration and adhesionCell Adhes. Migr., 5 (2011), pp. 11-15
- 59
- R.G. Hodge, A.J. RidleyRegulating Rho GTPases and their regulatorsNat. Rev. Mol. Cell Biol., 17 (2016), pp. 496-510
- 60
- W.A. Johnston, S.V. Moradi, K. AlexandrovAdaption of the leishmania cell-free expression system to high-throughput analysis of protein interactionsMethods Mol. Biol., 2025 (2019), pp. 403-421
- 61
- F. Cunningham, P. Achuthan, W. Akanni, J. Allen, M.R. Amode, I.M. Armean, R. Bennett, J. Bhai, K. Billis, S. Boddu, et al.Ensembl 2019Nucleic Acids Res., 47 (D1) (2019), pp. D745-D751
- 62
- F. Hormozdiari, M. van de Bunt, A.V. Segre, X. Li, J.W.J. Joo, M. Bilow, et al.Colocalization of GWAS and eQTL signals detects target genesAm. J. Hum. Genet., 99 (2016), pp. 1245-1260
- 63
- K.C. Cotto, A.H. Wagner, Y.Y. Feng, S. Kiwala, A.C. Coffman, G. Spies, A. Wollam, N.C. Spies, O.L. Griffith, M. GriffithDGIdb 3.0: a redesign and expansion of the drug-gene interaction databaseNucleic Acids Res., 46 (D1) (2018), pp. D1068-D1073
- 64
- E. Piñeiro-Yáñez, M. Reboiro-Jato, G. Gómez-López, J. Perales-Patón, K. Troulé, J.M. Rodríguez, H. Tejero, T. Shimamura, P.P. López-Casas, J. Carretero, et al.PanDrugs: a novel method to prioritize anticancer drug treatments according to individual genomic dataGenome Med., 10 (2018), p. 41
- 10
Lead contact
https://www.sciencedirect.com/science/article/pii/S0960982221007946?fbclid=IwAR2Fwcm8_8qxwe3QohG66zZXshEvsjT-9mpreQwqjOwCVzxA8Z0IyaSVt-E
..
Không có nhận xét nào:
Đăng nhận xét
Khi sử dụng tiếng Việt, bạn cần viết tiếng Việt có dấu, ngôn từ dung dị mà lại không dung tục. Có thể đồng ý hay không đồng ý, nhưng hãy đưa chứng lí và cảm tưởng thực sự của bạn.
LƯU Ý: Blog đặt ở chế độ mở, không kiểm duyệt bình luận. Nếu nhỡ tay, cũng có thể tự xóa để viết lại. Nhưng những bình luận cảm tính, lạc đề, trái thuần phong mĩ tục, thì sẽ bị loại khỏi blog và ghi nhớ spam ở cuối trang.
Ghi chú (tháng 11/2016): Từ tháng 6 đến tháng 11/2016, hàng ngày có rất nhiều comment rác quảng cáo (bán hàng, rao vặt). Nên từ ngày 09/11/2016, có lúc blog sẽ đặt chế độ kiểm duyệt, để tự động loại bỏ rác.